What are CCDS?
Cerebral Creatine Deficiency Syndromes (CCDS) are a group of inborn errors of creatine metabolism including AGAT, CTD, and GAMT. Symptoms may include: intellectual delays, expressive speech and language delay, autistic-like behaviour, hyperactivity, seizures, projectile vomiting in infancy, failure to thrive, and movement disorders.
Creatine helps supply energy to all cells in the body. It helps increase adenosine triphosphate (ATP).
Creatine is produced in the liver, which makes it out of three amino acids: arginine, glycine and methionine. Most of our body’s creatine (approximately 95%) is stored in the muscles that support the skeleton.
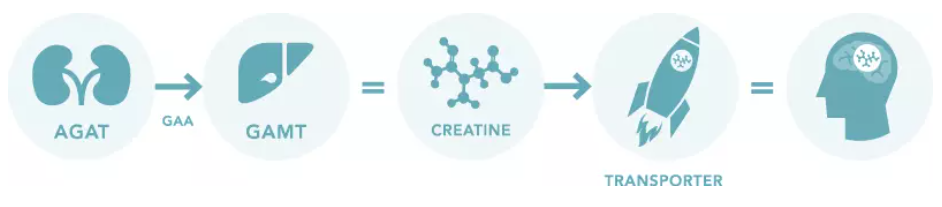
Normal Creatine Production Pathway:
Creatine is essential to sustain the high energy levels needed for muscle and brain development.
Creatine is created and delivered to the cells of the body in a three step process:
The AGAT enzyme breaks down arginine from the diet, producing GAA.
The GAMT enzyme utilizes GAA to form creatine.
Creatine Transporters move creatine into the cells of the brain and muscles for use.
Creatine is necessary to increase adenosine triphosphate (ATP), providing energy to all the cells in the body.
Cerebral Creatine Deficiency Syndromes
Cerebral Creatine Deficiency Syndromes (CCDS) are inborn errors of metabolism, which interrupt the formation or transportation of creatine. There are three CCDS and their pathways are shown below.
L-Arginine: Glycine Amidinotransferase (AGAT) Deficiency
Mutations found in the GATM gene result in AGAT Deficiency.
Patients with AGAT Deficiency lack the first enzyme necessary for creating creatine.
Treatment with oral supplementation of creatine monohydrate is effective in replenishing the body’s needed creatine supply and greatly improves outcomes if initiated early for AGAT Deficiency patients. Diet restrictions are not typically recommended for AGAT patients.
AGAT patients typically have less severe symptoms in comparison to GAMT and CTD because they have functioning transporters to utilize creatine found naturally in the diet and they do not have a buildup of GAA.
Guanidinoacetate Methyltransferase (GAMT) Deficiency
Mutations in the GAMT gene cause GAMT Deficiency.
Due to a lack of the GAMT enzyme, patients with GAMT are unable to break down the GAA formed in the first step of creatine synthesis. This results in a buildup of guanidinoacetate (GAA) which is believed to be toxic to the brain at high levels.
Treatment is aimed at reducing GAA production and supplying the creatine that is not produced by the body. Patients are typically prescribed oral supplements of creatine monohydrate and l-ornithine. Some physicians also recommend diet restrictions and sodium benzoate supplementation to further minimize GAA accumulation.
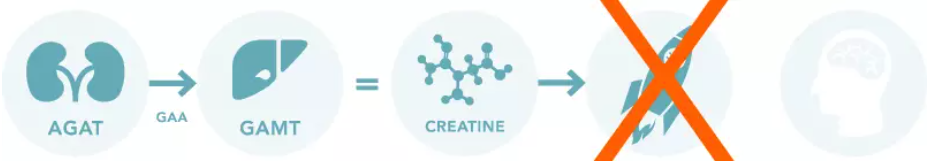
Creatine Transporter Deficiency (CTD)
CTD is also referred to as SLC6A8 Deficiency, CRTR, and X-linked Creatine Transporter Deficiency.
Mutations in the SLC6A8 gene result in CTD.
While patients with CTD have the necessary AGAT and GAMT enzymes to form creatine, the creatine transporter does not function properly. This results in creatine in the bloodstream, but not in the brain and muscles.
CTD patients do not experience high levels of GAA like GAMT patients and are not typically prescribed a restricted diet.
To date, there is no proven treatment for CTD. Visit our Research page to learn more about our commitments to help find a cure for CTD!